


 收藏
收藏 已收藏
已收藏作者:陈妍洁(主治医师 复旦大学附属中山医院消化内科)
指导者:沈锡中(主任医师 复旦大学附属中山医院消化内科)
1.糖原贮积病(glycogen storage disease,GSD)是一种遗传性疾病,主要病因为先天性糖代谢酶缺陷所造成的糖原代谢障碍,美欧国家的发病率为1/25000万~1/2000万。因糖原分解与合成有关的某些酶系统缺乏,糖原分解困难,糖原异常沉积于全身各组织,尤其是肝脏、心脏及肌肉中。
2.由于缺陷的酶不同,临床上分为13型。其中,Ⅰ、Ⅲ、Ⅳ、Ⅵ型以肝脏病变为主,Ⅰ、Ⅲ和Ⅳ型的肝脏损害最为严重,Ⅱ、Ⅴ、Ⅶ型则以肌肉组织受损为主。
3.Ⅰ型糖原贮积病又称肝型糖原累积病,临床最常见,由于缺乏葡萄糖-6-磷酸酶,不能将6-磷酸葡萄糖水解为葡萄糖。主要表现:①空腹诱发严重低血糖,患儿出生后即出现低血糖,惊厥以至昏迷。长期低血糖影响脑细胞发育,智力低下,多于2岁内死亡。②伴酮症和乳酸性酸中毒。③高脂血症,臀和四肢伸面有黄色瘤。向心性肥胖,腹部膨隆,体形呈“娃娃”状。④高尿酸血症。⑤肝细胞和肾小管上皮细胞大量糖原沉积。新生儿期即出现肝大,肾脏增大。成人后,可出现单发或多发肝腺瘤,进行性肾小球硬化、肾衰竭。⑥生长迟缓,形成侏儒状态。
4.临床表现为肝大、空腹低血糖、身材矮小、肥胖等。血生化检查:空腹血糖低,血甘油三酯及胆固醇水平升高,血乳酸、尿酸水平升高。胰高糖素试验:胰高糖素0.5mg肌内注射,每15min测血糖,持续2h,正常人10~20min后空腹血糖可上升3~4mmol/L,本病患者上升<0.1mmol/L,2h内血糖仍不升高,乳酸上升3~6mmol/L,并加重已有的乳酸性酸中毒,血pH降低。肝穿刺活检是本病确诊依据。测定患者肝糖原常超过正常值6%,葡萄糖-6-磷酸酶活性降低以至缺失,细胞核内有大量糖原沉积。果糖或半乳糖转变为葡萄糖试验:迅速静脉输注果糖(0.5g/kg)或半乳糖(1g/kg)配制的25%溶液,每10分钟取血1次,共1h,测定血葡萄糖、乳糖、果糖、半乳糖含量,患者血葡萄糖不升高,而乳酸明显上升。骨骼X线检查:可见骨骺出现延迟及骨质疏松。
5.本病预后不良,患者多在2岁之内夭折。2岁后生存者可出现智能低下,若4岁智能无明显减退者可望继续生存,代谢紊乱可望逐步纠正。未经正确治疗的本病患儿因低血糖和酸中毒发作频繁常有体格和智能发育障碍。伴有高尿酸血症患者常在青春期并发痛风或高血脂。如有痛风性关节炎、痛风石,可用别嘌醇和碳酸氢钠片加丙磺舒治疗。患者在成年期的心血管疾病、胰腺炎和肝脏腺瘤(或腺癌)的发生率高于正常人群,少数患者可并发进行性肾小球硬化症。
6.治疗用高蛋白、高葡萄糖饮食,多次喂养,以维持血糖正常水平,尤应于午夜加餐1次,以避免次晨低血糖。其他治疗包括防止感染,纠正酸中毒(可用NaHCO3,禁用乳酸钠)。纠正低血糖后如果血脂仍继续升高,可用氯贝丁酯(安妥明)50mg/(kg·d)。高尿酸血症如采用饮食疗法不能控制时,可用别嘌醇5~10mg/(kg·d)。激素治疗有益于维持正常血糖水平、提高食欲。胰高血糖素、各种类固醇激素、甲状腺素对改善症状皆可有暂时的疗效外科方法如做门-腔静脉吻合术,使肠吸收的葡萄糖越过肝,直接进入血液循环,可能术后肝缩小,生长加速,但长期效果并不肯定。亦有报告做肝移植者,效果不明且不易推广。其他有采用酶替代治疗等,但效果并不佳。
7.妊娠14~16周做宫内穿刺和羊水细胞培养测定其酸性麦芽糖活性,若见降低,则应中止妊娠。可通过胎儿肝活检测定葡糖-6-磷酸酶活力进行,通常在孕18~22周进行。预防措施包括避免近亲结婚,推行遗传咨询、携带者基因检测及产前诊断和选择性人工流产等。
1.患者基本情况
患者:女性,19岁,汉族,未婚,上海人。
入院时间:2016年4月8日。
主诉:反复关节肿痛伴肝功能异常1年。
现病史:2015年起患者出现左踝关节红肿疼痛,自服解热镇痛药3~4d后缓解。2016年初至今先后出现左踝关节、左手掌指关节及双侧肘关节红肿疼痛,体温波动于37.2℃左右,当地医院予解热镇痛药、碳酸氢钠片、秋水仙碱治疗后症状均可缓解。伴有反复轻度肝功能异常。患者诉每次发病均于活动后出现,且自幼即感活动耐力差、易乏力、易饥饿。
个人史:10岁时患有传染性单核细胞增多症。幼年湿疹病史。15岁初潮,月经周期正常,否认痛经史。
家族史:其母亲与外公均有血小板增高症;父亲有痛风病史。
2.入院查体
一般生命体征:T 37.5℃,P 78次/min,R 20次/min,BP 110/64mmHg。智力低下,对答尚切题;身高150cm,体重40kg,体形消瘦,发育迟缓,营养状态差,头发稀疏;心肺无特殊;肝右侧肋下4横指,质韧,表面光滑边缘钝;四肢脊柱无畸形,四肢可见少许色素沉着,右肘关节稍肿,踝关节、跖趾关节及趾骨间关节未见异常。四肢肌力、肌张力正常,病理征(-)。
3.入院辅助检查
血红蛋白105g/L;血小板计数414×109/L;白细胞计数6.9×109/L;尿蛋白(+)~(+++);尿红细胞(-);谷丙转氨酶44U/L;谷草转氨酶60U/L;碱性磷酸酶108U/L;γ-谷氨酰转移酶90U/L;血尿素1.5mmol/L;血肌酐 31μmol/L;估算肾小球滤过率>120mL/(min·1.73m2);血尿酸612μmol/L;24h尿液蛋白定量0.86g;尿免疫球蛋白10.9mg/L;尿转铁蛋白21.43mg/L;24h尿白蛋白563.6mg;尿白蛋白281.8mg/L;血糖4.1mmol/L;糖化血红蛋白4.8%;血乳酸6.60mmol/L;总胆固醇6.50mmol/L;甘油三酯7.04mmol/L;血沉91mm/h;C反应蛋白84.8mg/L;消化道肿瘤标志物、风湿免疫、自身抗体、甲状腺功能、性激素水平等指标均未见异常。
4.初步诊断思维过程
(1)入院时病情总结
①19岁女性,因“反复关节肿痛伴肝酶升高1年”入院;②近1年先后出现活动后中小关节、非对称性的红肿痛,无肌肉症状,解热镇痛药、碳酸氢钠片、秋水仙碱治疗有效,自幼活动耐力差、易饥饿;③15岁初潮,月经周期正常,否认痛经史。其母亲与外公均有血小板增高症;父亲有痛风病史;④体格检查:智力低下,体形消瘦,营养状态差,发育迟缓,肝右侧肋下4横指,质韧,表面光滑边缘钝,右肘关节稍肿;⑤实验室检查肝酶轻度升高,血乳酸升高,尿蛋白(+)~(+++),24h尿液蛋白定量0.86g,血尿酸、总胆固醇、甘油三酯增高,血沉增快,C反应蛋白增高。
(2)入院时诊断思路
患者以反复关节肿痛伴肝功能异常入院,目前存在多方面代谢紊乱表现为关节红肿疼痛、高尿酸血症、脂质代谢紊乱、发育异常、活动耐力差、肝大。考虑患者肝功能异常伴肝大,需要进一步完善腹部影像学检查以明确诊断。
(3)入院初步诊断
①肝功能异常;②高尿酸血症;③关节炎?④脂质代谢紊乱;⑤发育异常;⑥活动耐力减退;⑦肝大。
5.后续检查
腹部B超:①脂肪肝;②肝多发实质占位;③胆囊壁胆固醇结晶沉着症;④双肾偏大,双肾海绵肾样改变。
肝脏MRI检查(图1):①肝脏多发占位:考虑腺瘤可能性大,最大一枚(肝右前叶下段)恶变不除外,请结合临床进一步检查;②肝脏弥漫性多发增生结节可能性大,随访;③肝大,脂肪肝。
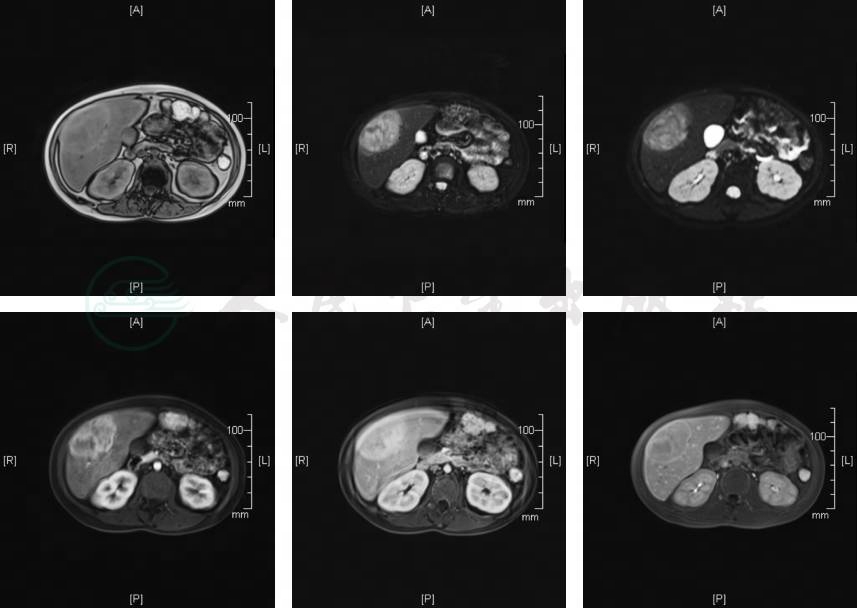
图1 肝脏MRI检查
第一排从左到右依次是T1W1、T2W1、DWI;第二排从左到右依次是增强动脉期、增强门脉期、增强肝胆特异期。
肝脏穿刺病理(图2):一条可见肝细胞胞浆空亮,胞膜清晰增厚,另一条汇管区不明显,可见肝细胞增生伴肝窦扩张充血,灶性纤维组织增生。免疫组化:肝型脂肪酸结合蛋白(liver-fatty acid binding protein,L-FABP)(-),CD34(血管+),CK19(少量+),C反应蛋白(+),Ki-67(5%阳性),GS(+),Hsp70(-),GPC3(-),B-cat(膜 +),SAA(-)。特殊染色:网染(网状纤维增生),过碘酸希夫(periodic acid-Schiff,PAS)染色(+)。
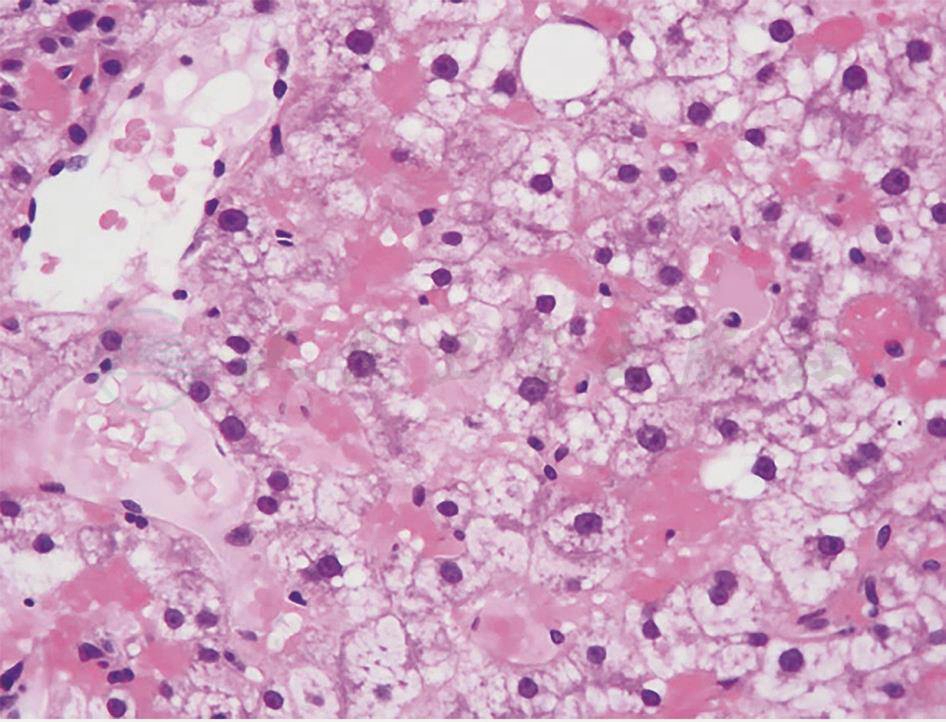
图2 肝脏穿刺病理(HE染色,100×)
6.最终诊疗思维过程
(1)最终诊断思路
糖原贮积病(glycogen storage disease,GSD)是一种遗传性疾病,主要病因为先天性糖代谢酶缺陷所造成的糖原代谢障碍,欧美国家的发病率为1/25000万~1/2000万。因某些糖原分解与合成相关酶系统缺乏,糖原分解困难,糖原异常沉积于全身各组织,尤其是肝脏、心脏及肌肉中。由于缺陷的酶的不同,临床上分为13型。其中Ⅰ、Ⅲ、Ⅳ、Ⅵ型以肝脏病变为主,Ⅰ、Ⅲ和Ⅳ型的肝脏损害最为严重;Ⅱ、Ⅴ、Ⅶ型则以肌肉组织受损为主。
Ⅰ型糖原贮积病主要表现:①空腹低血糖,胰高血糖素治疗无效。②肝大。③高脂血症:高甘油三酯血症和高脂肪酸血症,可沉积于臀和四肢伸面形成黄瘤。④伴有酮症、乳酸性酸中毒、高尿酸血症。⑤肾脏受累,逐渐表现为蛋白尿、高血压和终末期肾病;由于肾脏糖原聚集,肾脏增大。⑥患者成年后多数身材矮小,代谢控制不佳者可出现单发或多发肝脏腺瘤、痛风、骨质疏松、尿石症等晚期并发症。这些症状均与本例患者相符。结合患者肝穿刺病理提示大量糖原沉积,PAS染色(+),因此该患者考虑诊断糖原贮积症(Ⅰ型)。进一步可行果糖或半乳糖转变为葡萄糖试验、葡萄糖-6-磷酸酶 (glucose-6-phosphatase,G-6Pase)活性测定及基因检查,与其他型进行鉴别。
(2)鉴别诊断
肝恶性肿瘤、转移性肝恶性肿瘤、自身免疫性疾病(系统性红斑狼疮、混合性结缔组织病等)。
(3)最终诊断
糖原贮积症(Ⅰ型),继发性高尿酸血症,高脂血症。
(4)治疗方案
1)一般治疗
少量多餐饮食防止低血糖休克或酸中毒的发生。
2)药物治疗
维生素类药物,如B族维生素、维生素C等。
3)并发症的治疗
有感染给抗生素治疗。
4)手术治疗
做门-腔静脉吻合术,改善本病的血生化异常。
本例为罕见病,为遗传性疾病,对该病认识不足是引起误诊、漏诊的主要原因。
[1]KISHNANI PS,AUSTIN SL,ABDENUR JE,et al.Diagnosis and management of glycogen storage disease type I:a practice guideline of the American College of Medical Genetics and Genomics[J].Genet Med,2014,16(11):e1.
[2]MUNDY HR,HINDMARSH PC,MATTHEWS DR,et al. The regulation of growth in glycogen storage disease type 1[J].Clin Endocrinol (Oxf),2003,58(3):332-339.
[3]DUNGER DB,HOLDER AT,LEONARD JV,et al. Growth and endocrine changes in the hepatic glycogenoses[J].Eur J Pediatr,1982,138(3):226-230.







