


 收藏
收藏 已收藏
已收藏英文名称 :antineutrophil cytoplasmic antibody associated vasculitis
ANCA相关性血管炎 (antineutrophil cytoplasmic antibody associated vasculitis,AAV)是一组累及小至中等血管 (小动脉、毛细血管、小静脉)的系统性血管炎,血清中存在靶抗原为蛋白酶3(PR3)或髓过氧化物酶 (MPO)的抗中性粒细胞胞浆抗体 (ANCA)。AAV主要包括显微镜下多血管炎 (microscopic polyangiitis,MPA),肉芽肿性多血管炎 [granulomatosis with polyangiitis,GPA,旧称韦格纳肉芽肿 (Wegner granulomatosis,WG)]和嗜酸性肉芽肿性多血管炎 [Eosinophilic granulomatosis with polyangiitis,EGPA,旧称变应性肉芽肿性血管炎或 Churg-Strauss综合征(CSS)]。它们累及的血管和临床表现有很多相似之处,大多累及多个器官或系统,可隐匿或缓慢起病,也可急性或暴发性发病。不同AAV又有其各自的临床和病理特点:
GPA是以毛细血管、微小动静脉受累为主的系统性、坏死性、肉芽肿性血管炎。典型的GPA三联征包括上呼吸道、下呼吸道 (肺)及肾脏病变。
MPA是一种主要累及小血管的系统性、坏死性血管炎,可侵犯肾脏、皮肤和肺等脏器的小动脉、微动脉、毛细血管和微小静脉。常表现为坏死性肾小球肾炎和肺毛细血管炎。
EGPA是一主要累及中、小动脉和静脉的系统性、坏死性血管炎,病理特征为受累组织有大量嗜酸性粒细胞浸润和血管外肉芽肿形成及坏死性血管炎。
目前我国AAV的发病率仍不明确。近20年来AAV的发病率逐渐升高,欧洲和北美的年发病率约为20~30/100万,GPA和MPA的现患病率分别为130/100万和47.9/100万。男性略多于女性,MPA的男女患病比率为(1~1.8)∶1。可见于从儿童到老年人的任何年龄段,以老年人多见,50~70岁为发病高峰。地域对于发病率有影响,北欧地区的MPA发病率是美国的5倍。
ANCA相关性血管炎的病因尚不清除,但目前认为是具有某种遗传倾向的患者在环境诱因(如感染、毒物和药物)的作用下发生的一种自身免疫性疾病。多种炎症细胞和因子参与起病。细胞因子介导的黏附分子的表达和功能异常,以及白细胞和血管内皮细胞的异常激活在MPA的发病中可能都起一定作用,ANCA可能在AAV的发病中起一定作用。但具体的启动因素尚不清楚。可能的发病机制有:
1.遗传因素
GPA可能和HLA-B50、B55、DR1、DR2、DR4、DR8、DR9和DQw7有关,确切的关系还有待进一步研究。
2.环境因素
研究表明,GPA可能和病毒(EB病毒、巨细胞病毒和细小病毒B19)感染、细菌(金黄色葡萄球菌、革兰阴性杆菌)感染及接触硅物质等有关。变应性鼻炎和哮喘在EGPA患者中很常见,可能与吸入或接触某些特殊的过敏原或化学物质有关。
3.抗中性粒细胞胞浆抗体
1985年VanderWoude等首先提出ANCA与GPA有相关性,此后许多研究都肯定了ANCA与三种主要的AAV均密切相关。根据ANCA的免疫荧光检测结果可分为两种染色型:胞浆型(c-ANCA)和核周型(p-ANCA)。已经发现了导致这些荧光染色型且与系统性血管炎密切相关的抗原,即c-ANCA对应蛋白酶3(PR3)而p-ANCA则对应髓过氧化酶(MPO)。抗髓过氧化物酶(MPO)抗体和抗蛋白水解酶3(PR3)抗体可能参与了ANCA相关性血管炎的发生。70%的MPA患者存在p-ANCA,在MPA患者中结构性表达PR3的中性粒细胞比例升高,这些中性粒细胞可被ANCA直接激活而不用预先用抗原活化。在体外试验中,ANCA可与中性粒细胞和单核细胞中的颗粒结合或通过膜上的Fc端结合,激活已活化的中性粒细胞使之释放活性氧自由基和溶酶体酶,导致内皮细胞的破坏和溶解,从而引起血管炎症和坏死。活化的中性粒细胞黏附并破坏血管内皮细胞的同时还可以吸引更多的中性粒细胞,从而在微环境中产生自放大反馈路径。在体内试验中,用鼠髓过氧化酶免疫髓过氧化酶缺限型小鼠的脾细胞转入野生型小鼠体内可以诱发新月体肾炎等寡免疫性系统性血管炎。然而在PR3缺陷型小鼠上进行类似的试验不能引起明显的血管炎病变。说明MPO-ANCA与PR3-ANCA可能通过不同的机制参与发病。
目前关于ANCA致AAV发病机制推测如下:感染性促发因子或其他环境刺激因素导致细胞因子释放,进而致敏中性粒细胞或单核细胞,导致ANCA抗原在细胞表面表达,被激活的中性粒细胞黏附在内皮细胞表面,ANCA与其抗原结合,导致中性粒细胞激活,释放出活性氧族和溶酶体酶,进一步导致内皮细胞损伤并活化内皮细胞表面。ANCA还可通过中性粒细胞表面的Fcγ受体相结合。脱颗粒炎症介质与内皮细胞结合并作为ANCA靶点。同时趋化细胞因子如IL-8,MCP-1和黏附分子一起,放大化学趋化和炎症细胞迁移,通过激活旁路补体途径,导致炎症进一步扩增。最终导致严重的血管壁坏死性炎症。
4.内皮细胞抗体
在ANCA相关血管炎的病程早期,内皮细胞可募集炎症细胞并促使它们在血管损伤部位黏附。PR3和其他中性粒细胞蛋白酶的释放可诱导内皮细胞合成和分泌IL-8,后者是一种趋化因子,可以募集更多的中性粒细胞。可溶性内皮细胞蛋白C受体通过与PR3相互作用而与活化的中性粒细胞结合,这一机制联系了粒细胞活化、血管损伤和凝血三个方面,也为MPA患者和GPA患者容易发生静脉血栓的现象提供可能的解释。还有研究证据显示器官特异性抗内皮细胞抗体(AECA)在发病中起一定作用,通过和血管内皮细胞表面抗原结合从而导致细胞损伤和血管炎症。
5.B细胞
B淋巴细胞在ANCA相关血管炎的发病中可能起重要作用。研究发现循环中活化的B淋巴细胞数与疾病活动性评分具有相关性,在AAV活动期患者中B淋巴细胞刺激因子水平明显升高。ANCA相关性血管炎应用B细胞去除治疗取得了较为乐观的结果,为ANCA相关血管炎的病理生理学机制提供新的视角。B细胞去除治疗有效的机制还不清除,但可能与ANCA的产生减少、B淋巴细胞抗原呈递和产生细胞因子功能减弱有关。
6.T细胞和细胞因子
体外实验发现,PR3特异性的CD4+Th17细胞在ANCA相关性血管炎病灶中有过度表达。临床研究表明,ANCA相关性血管炎体内各型T细胞比例失调,CD8+T细胞的转录特征可预测该疾病的复发几率。
GPA的典型病理表现可见小血管的纤维素样变性,血管壁中性粒细胞浸润,坏死性肉芽肿形成。但多数病例仅可见血管炎、肉芽肿和坏死三种改变中的一种或两种。GPA的肾脏病理改变为局灶节段性肾小球肾炎,免疫组化显示寡/无免疫复合物沉积。
MPA典型的组织病理学特点是肺毛细血管炎。在此病变中,肺泡间质组织断裂,肺组织毛细血管网的完整性遭到破坏,导致红细胞漏入肺泡内。肺泡壁水肿形成纤维素样坏死。肺泡隔内有明显的中性粒细胞浸润伴白细胞碎裂现象是其特征性病理改变。免疫荧光检查很少或没有免疫复合物沉积。其他表现包括毛细血管血栓形成、Ⅱ型肺泡上皮细胞过度增生和淋巴细胞及浆细胞浸润。病情严重者可出现弥漫性肺泡出血。肾脏病理改变是坏死性肾小球肾炎,其特点是节段性坏死,新月体形成(毛细血管外增生),毛细血管内增生轻微或缺如。与肺脏受累相似,肾脏免疫荧光检查很少或无免疫复合物沉积,电镜下未见或仅有轻微的高电子密度沉积物。此病变与免疫复合物介导的肾小球肾炎和抗肾小球基底膜(GBM)抗体介导的疾病有显著区别,但与GPA或特发性急进性新月体肾小球肾炎病变难以区别。肌肉和腓肠神经活检可见小到中等静脉的坏死性血管炎。
EGPA特征性的病理变化包括坏死性血管炎、嗜酸性粒细胞组织浸润和血管外性肉芽肿。
1.常规检查
急性期炎症指标如ESR、CRP升高,部分患者有贫血、白细胞和血小板增多。累及肾脏时出现蛋白尿、镜下血尿和红细胞管型尿,血清肌酐和尿素氮水平升高。EGPA患者的外周血嗜酸性粒细胞增多,一般在1.5×109/L以上,同时血清中IgE升高,病情缓解后可下降。
2.自身抗体
ANCA是本组疾病的血清学标记,见于90%以上病情活动的GPA和MPA患者,以及约30%的EGPA患者,是明确诊断、监测病情活动和预测复发的重要指标。ANCA按其免疫荧光类型可分为p-ANCA和c-ANCA,p-ANCA为核周型,其主要靶抗原为髓过氧化物酶(MPO);c-ANCA为胞浆型,靶抗原为蛋白水解酶3(PR3)。PR3-ANCA对活动性GPA的诊断有较高敏感性及特异性。MPO-ANCA主要见于MPA和EGPA,肺受累及者常有此抗体。ANCA对于鉴别MPA和典型的结节性多动脉炎(PAN)很有帮助,后者是一种主要累及中等肌性动脉的血管炎。
约40%的患者可查到抗心磷脂抗体,少部分人抗核抗体、类风湿因子阳性。抗肾小球基底膜(GBM)抗体相关疾病比ANCA相关性血管炎更为少见,但也可表现为肺-肾综合征,而且两者可同时发生,10%~40%的抗GBM阳性的患者也存在ANCA,其中少部分患者同时存在系统性血管炎的表现。
3.影像学改变
胸部X线和高分辨CT检查对AAV的诊断和鉴别诊断非常重要。GPA患者常显示双肺多发性浸润和结节病灶,常有空洞形成,以双下肺多见;X线和CT可显示鼻窦黏膜增厚,甚至鼻窦骨质破坏。MPA患者在早期表现为无特征性肺部浸润影或小泡状浸润影,双侧不规则的结节片状阴影,肺空洞少见,中晚期可出现肺间质纤维化。EGPA患者以多变性肺部阴影为特点,多数患者呈现肺内浸润性病变,可呈结节状、间质性或斑片状阴影,阴影可迅速消失。GPA和EGPA还可出现胸腔积液,EGPA患者的胸腔积液富含嗜酸性粒细胞。DAH的X线表现无特异性,显示片状至弥漫性的肺泡浸润,可见于MPA和GPA患者。
4.病理检查
受累器官的活检病理是诊断和鉴别诊断的关键。即使存在提示血管炎的临床表现和ANCA阳性,在给予患者长期有潜在毒性作用的药物治疗之前应尽可能寻找病理学依据来证实临床诊断。
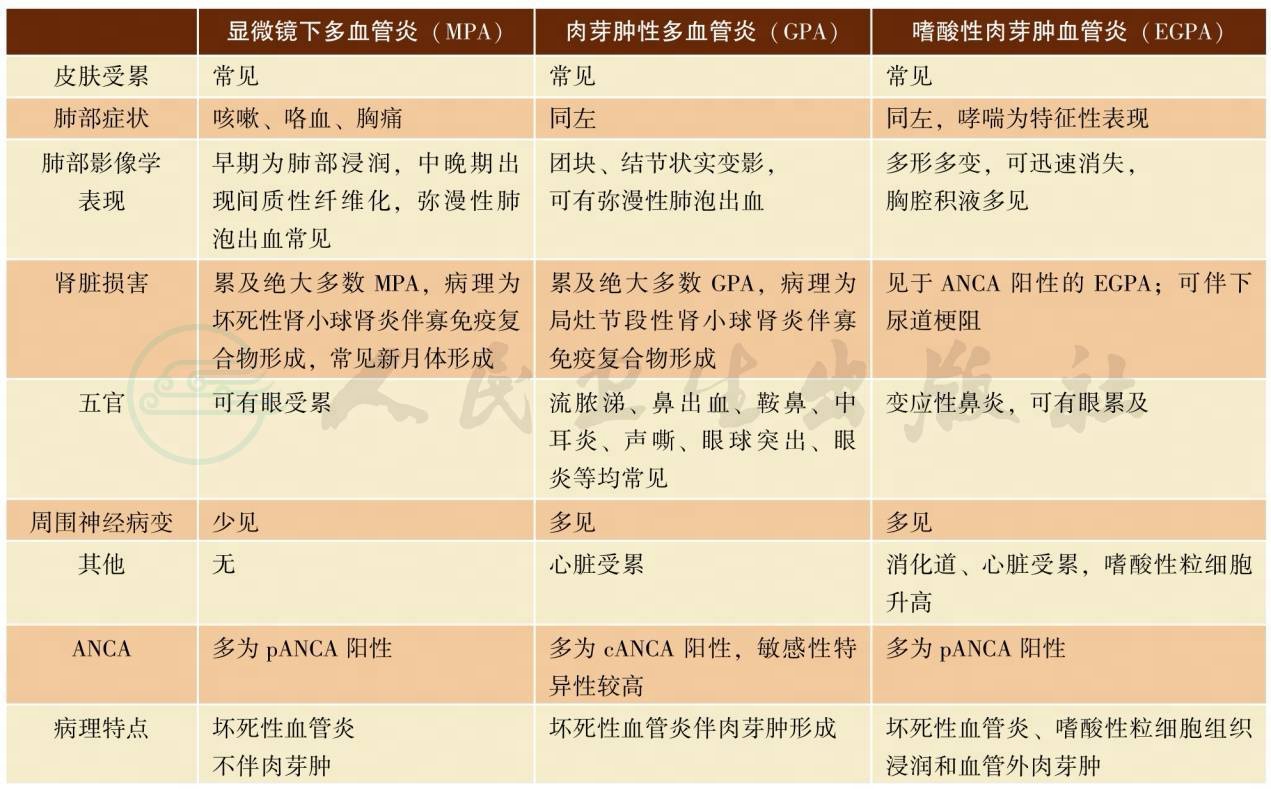
上呼吸道、支气管内膜及肾脏活检是诊断GPA的重要依据,典型病例可见小血管的纤维素样变性,血管壁中性粒细胞浸润,坏死性肉芽肿形成。但多数病例仅可见血管炎、肉芽肿和坏死三种改变中的一种或两种。GPA的肾脏病理改变为局灶节段性肾小球肾炎,免疫组化显示寡/无免疫复合物沉积。MPA普遍存在肾损害,病例表现为坏死性肾小球肾炎,特征为节段性坏死、新月体形成、轻/无毛细血管内增生,免疫组化同样为寡/无免疫复合物沉积。EGPA特征性的病理变化包括坏死性血管炎、嗜酸性粒细胞组织浸润和血管外性肉芽肿。皮肤活检多显示为白细胞碎裂性血管炎,免疫荧光镜检很少或没有补体和免疫球蛋白沉积(表1)。
表1 MPA、GPA和EGPA的临床特点比较
对于肾活检或皮肤活检未发现提示性病理特征时,可考虑行肺活检,通常采用开胸肺活检或经胸腔镜肺活检(VATS)。不足10%的患者可通过经支气管镜肺活检获得的少量标本得出明确诊断。
AAV病情复发的比例非常高,MPA患者中约占30%。文献中报道的复发率范围很宽,可能与治疗方案和随访时间不同有关。在CYCAZAREM试验中,所有患者均接受积极的CTX治疗,其复发率在18个月仅有15%。在严密的随诊过程中很少发生严重的病情复发。
1.病情复发的危险因素
感染可诱导ANCA抗原在循环中性粒细胞表达导致中性粒细胞脱颗粒、释放氧自由基和血管损伤,因此感染一直被认为是诱发病情活动的因素之一。激素减量过快或过早停用也可能是病情复发的诱因之一。在现有的3项ANCA相关血管炎的大型随机临床试验中,CYCAZAREM试验的病情复发率最低,其激素治疗的疗程也最长(达18个月)。如果患者先前有过病情复发,则再次复发的风险明显高于无复发病史的患者。ANCA对于预示病情复发亦有重要作用,临床缓解一段时间后ANCA仍持续阳性者,或由阴性转为阳性者,或ANCA滴度进行性升高者预示病情复发的可能性较大。
2.监测ANCA滴度
血浆ANCA滴度大多与血管炎的病情活动度相平行。在一项前瞻性随机临床试验中,入选20名无临床症状但近期ANCA滴度升高的患者,9名患者给予预防性激素和CTX治疗,其余则仅定期随诊。预防治疗组无一例出现临床病情活动,但未治疗组有9例出现病情活动,多出现于ANCA滴度升高后数月。然而,亦有约三分之一的患者不存在这种平行关系。在一项对60名病情缓解期患者(80%ANCA阴性)的观察性研究中,23名患者出现病情活动,其中13例先出现ANCA滴度升高,但亦有6例ANCA滴度升高的患者临床并未出现病情活动。在另一项100例患者的病例队列研究中,29%的患者虽有ANCA滴度升高但无病情活动。因此,不建议单纯依赖ANCA滴度来指导治疗策略的制订,但在病程的第1年应每月监测ANCA,对于ANCA滴度升高的患者应加强临床监测。
3.病情复发的临床识别
大多数复发出现在停止免疫抑制剂治疗后5年之内,复发时累及的脏器可以与初发时的受累脏器相同或不同。虽然病情复发与初发累及的器官大多相同,但在评价病情复发时明确新发症状是否因病情活动所致非常重要。对于存在全身炎症性症状和同样的脏器受累症状者,临床判断比较容易。但是对于另一些患者可能需要进行组织活检以发现明确的血管炎证据并除外感染和非炎症性病情进展。机会性感染是特别需要进行鉴别的重点,因为即使停用的激素和CTX治疗,患者的免疫功能抑制状态仍会持续一段时间。病情复发活动导致的晚期进行性肾衰竭并不少见,但首先需要除外修复期肾小球缺血和硬化导致的血管腔狭窄以及肾单位丢失继发的健存肾单位血流动力学和代谢紊乱(如肾小球高滤过压)造成的肾功能恶化。此类患者的血清肌酐大多上升缓慢,尿检有蛋白尿,但无活动性尿沉渣,也无其他提示病情活动的临床症状。还有些患者可能是合并了血管炎以外的肾脏病变,如IgA肾病。







