


 收藏
收藏 已收藏
已收藏英文名称 :autosomal dominant polycystic kidney disease
中文别名 :成人型多囊性肾脏病
常染色体显性遗传多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)曾被称为成人型多囊性肾脏病,是最常见的多囊肾病,具有遗传异质性。全球发病率为1/(1000~2500)。男女罹患机会无明显差异。主要表现为肾脏囊肿的发生、增多和增大,多系统受累。本病在严重程度、进展至终末期肾病的时间及肾外表现等方面的个体差异很大,甚至体现在同一家系中。
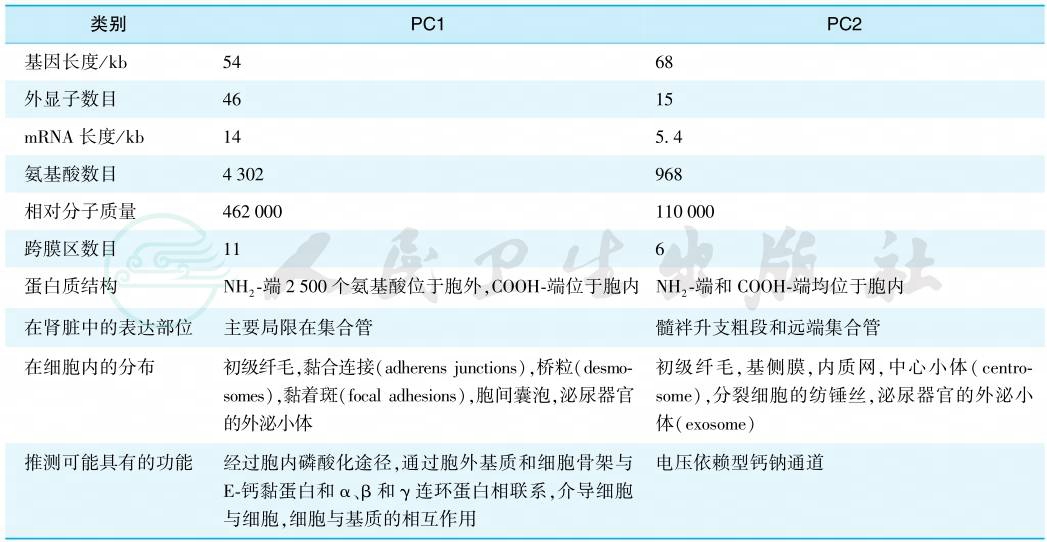
PKD1和PKD2两个基因突变可致ADPKD。PKD1基因位于16号染色体(16p13.3),编码多囊蛋白1(polycystin1,PC1),占ADPKD患者的85%。PKD2基因位于4号染色体(4q21),编码多囊蛋白2(polycystin2,PC2),约占患者中的15%。两种多囊蛋白的比较见表1。具有PKD2突变的患者肾功能损害进展稍慢。同时具有两者突变(transheterozygotes)的患者病变更加严重。还有极少数患者发现存在PKD3基因,但确切的异常位点尚未定位。目前为止,已经发现PKD1基因中有1923种截断突变,但主要发生在3’端。而5’端突变的患者病变更重,更容易伴有颅内血管瘤及血管瘤破裂出血。而在PKD2基因中,目前发现有241种类型各异的突变。
表1 两种多囊蛋白的比较
仅有1%左右的肾单位会演变成肾囊肿,以往认为患者从父母一方遗传了含有突变PKD1或PKD2的基因,从另一方遗传了野生型基因,这种先天存在的突变PKD1或PKD2基因影响到所有细胞,出生后少数细胞的正常等位基因又在感染、中毒等外界因素作用下发生体细胞突变(somatic mutation),即“二次打击模型(two-hit model)”,双重突变导致细胞正常功能丧失,后一次“打击”触发囊肿形成并决定其发生时间和部位。然而,PKD1或PKD2的单一等位基因不足会产生正常基因产物水平的随机波动,即便没有体细胞突变的“二次打击”,也会降低疾病产生的阈值(单一等位基因不足模型)。目前越来越多的证据支持单一等位基因不足模型。当然,PKD1和PKD2单倍体状态的基因不稳定性也会增加体突变二次打击的可能性,导致囊肿的形成和疾病的进展。
PC1和PC2通过细胞内的羧基端螺旋区相互连接作用,促进PC1转移到浆膜上,并稳定PC2的钙通道活性。PC1或PC2的缺乏或不足会导致细胞内钙离子浓度的下降,从而刺激环磷腺苷(cAMP)介导的囊肿上皮细胞的增生。
具有高增殖指数的肾小管的发育更容易受PC1和PC2水平减少的影响。而疾病的严重程度取决于基因灭活的时间。小鼠实验中如果PKD1和PKD2基因在尚未成熟的小管上皮细胞增生过程中灭活,就会引起巨大的囊肿及胚胎或新生儿期死亡。而如果基因灭活发生在肾脏上皮细胞已经分化成肾单位后,则病变较轻。
ADPKD患者早期肾脏大小正常,后期则增大,并出现形态异常,如肾盂肾盏的异形、肾乳头及肾锥体的完整结构受到破坏等。囊肿呈球形,大小不一。初起时肾内可仅有少数囊肿,随病程进展而渐增多,最终全肾均由囊肿所占,肾脏可达足球大小。光镜下,囊肿间尚可见到完整肾结构,从正常表现到肾小球硬化、小管萎缩、间质纤维化等程度不一,这些改变均为囊肿压迫引起肾缺血所致。电镜下显示的囊肿上皮细胞与近端或远端小管上皮细胞相似。囊液一般较清晰,当出现囊内感染或出血时则可为脓性或血性。







