


 收藏
收藏 已收藏
已收藏英文名称 :congenital esophageal atresia and tracheoesophageal fistula
中文别名 :食管闭锁-气管瘘
先天性食管闭锁及气管食管瘘(congenital esophageal atresia and tracheoesophageal fistula)简称“食管闭锁-气管瘘”,是一种严重的先天性发育畸形。发病率为1∶(3 000~4 000)个活产新生儿,早产未成熟儿多见。多达50%患儿伴有其他畸形,其中25%是危及生命或需急诊手术的,如肛门闭锁、肠旋转不良、肠闭锁等。先天性心脏病是最常见的合并畸形,其并发症发生率和死亡率也最高。其他常见的合并畸形包括泌尿生殖系统、骨骼、肛门、直肠和其他胃肠道,最多见十二指肠闭锁。畸形可单发,也可几种畸形同时存在,1973年Quan等用VATER综合征表示合并的畸形(V脊柱、A肛门直肠、TE气管食管瘘、R桡骨/肾脏)。以后扩展为 VACTERL(C心脏和L肢体)。
最早发现本病的是 Willion Durston医生,他于1670年报告了一对胸部连体婴中的一个食管近端盲闭。1697年,Hhomas Gibsin详细描述了一例食管闭锁-气管瘘患儿的基本特征。1939年第一次手术修复成功,Leven和Ladd各自报道了一例分期手术成功的病例。1943年Haight和Towsley报告了第一例一期吻合成功的病例。近年来,随着手术技术、麻醉和新生儿监护水平的提高,静脉营养及抗生素的合理使用,使患儿成活率不断提高,目前国际上体重1 500g以上,没有严重心脏畸形的患儿成活率已达97%以上。现在越来越关注患儿的术后并发症,长期临床预后与生活质量。
本病发病机制目前仍不清楚,有人认为与炎症、血管发育不良或遗传因素有关。通过家族中垂直以及横向散发的病例报道认为本病是多基因遗传疾病。研究发现,10%的病例表现有非特异性染色体异常,如易位、缺失和复制。胚胎期原始前肠在发育过程中,贯通和分隔发生障碍,就可形成食管闭锁和食管气管瘘。根据不同形态的畸形,可分为5种病理类型(图1)。
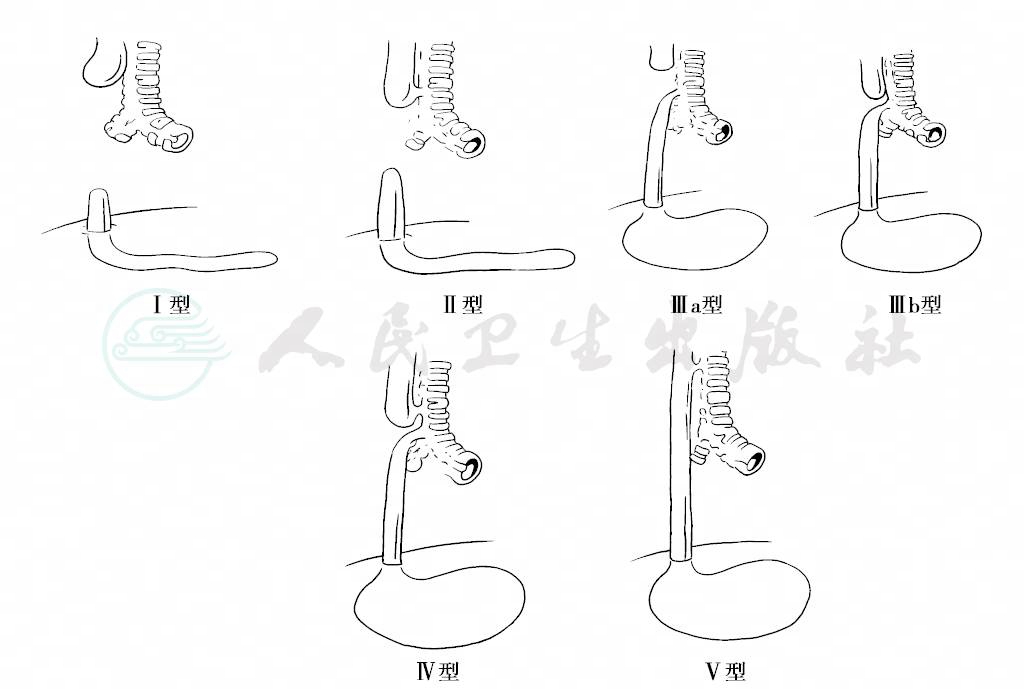
图1 食管闭锁分型
Ⅰ型:食管近、远端均为盲端,无食管气管瘘(4%~8%)。
Ⅱ型:近端食管有瘘管与气管交通,远端食管为一盲端(0.5%~1%)。
Ⅲ型:近端食管为一盲端,远端食管有瘘管与气管交通(85%~90%)。
Ⅳ型:食管近、远端各有瘘管与气管交通(1%)。
Ⅴ型:无食管闭锁,但有瘘管与气管交通,呈H型(4%~5%)。
以最常见的Ⅲ型为例。由于食管上段盲袋容量仅几毫升,不能吞咽的唾液反流入气管,引起吸入性肺炎或肺不张;由于存在食管下段与气管之间的瘘管,气体可经过气管食管瘘进入胃肠道引起腹胀,使膈肌抬高,同时有效潮气量减少,造成肺通气功能严重受损,瘘越大,腹胀越严重,呼吸困难就越严重;高酸度的胃分泌物通过气管食管瘘反流进入气管,使肺实质发生一种严重的化学刺激性肺炎。
1994年Spitz等认为影响预后的主要因素是体重和是否合并严重的先天性心脏病,提出了一个根据上述两点的简化的预后分级方法(表1)。
表1 Spitz分级方法








