


 收藏
收藏 已收藏
已收藏英文名称 :hereditary fructose intolerance
中文别名 :果糖血症
遗传性果糖不耐症是一种常染色体隐性遗传病,由于1-磷酸果糖缩醛酶缺失导致肝脏、肾脏以及小肠中果糖-1-磷酸累积。在一些欧洲国家的发病率为1/20 000。纯合子新生儿出生时健康,直到使用含果糖饮食,一般出现在断奶后开始增加果糖或蔗糖时起病。食用果糖或者其他可以代谢成1-磷酸果糖的糖类会出现包括严重腹痛、呕吐和低血糖症在内的临床症状。长时间的使用果糖会导致肝脏和(或)肾衰竭直至死亡。患者必须远离甜食。由于醛缩酶B缺陷导致无法将1-磷酸果糖剪切生成磷酸二羟丙酮和D-甘油醛。
正常情况下,外源性果糖通过空肠黏膜吸收后,在醛缩酶B的作用下形成甘油醛和磷酸二羟丙酮,进一步形成3-磷酸甘油醛,最终代谢为用于糖异生或糖酵解的1,6-二磷酸果糖,或者进入糖酵解途径,转化成丙酮酸盐,进入三羧酸循环。果糖代谢示意途径见图1。
醛缩酶B基因遗传性缺陷使醛缩酶B结构和活性发生改变,患者摄入或输注含果糖成分的物质后,1-磷酸果糖在肝中堆积,消耗细胞内库存的无机磷酸盐(Pi),造成三磷酸腺苷(ATP)缺乏,肝细胞ATP依赖性离子泵功能障碍,膜内外离子梯度不能维持,细胞肿胀,引起组织如肝脏、肾小管功能障碍,导致代谢平衡紊乱,阻碍糖原分解和糖异生,产生低血糖。持久的含果糖饮食会造成患儿肝细胞坏死、脂肪浸润、胆小管增生和纤维化甚至肝硬化。
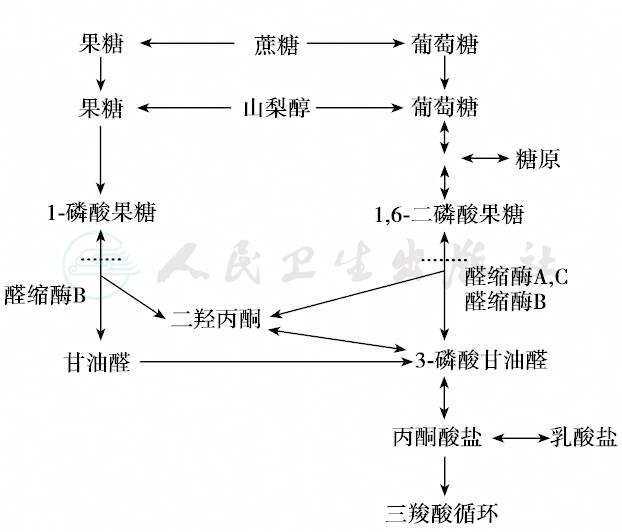
图1 果糖代谢途径
虚线表示醛缩酶B缺陷导致的代谢环节发生障碍
果糖广泛存在于各种水果和蔬菜中,因此人体自日常饮食中摄入的果糖量较大。果糖进入人体后大部分在肝脏中进行代谢,仅小量由肾小管和小肠代谢果糖作为人体摄入的另一种单糖。其体内代谢过程为:首先在肝肾及肠黏膜果糖激酶的催化下,转变为1-磷酸果糖;后者在1-磷酸果糖醛缩酶的作用下分解成磷酸二羟酮及甘油醛;后者通过甘油醛激酶磷酸化作用转变成磷酸甘油醛,再经果糖-1,6-二磷酸醛缩酶催化磷酸甘油醛与磷酸二羟丙酮缩合成1,6-二磷酸果糖。后者再由果糖-1,6-二磷酸酶水解成果糖-6-磷酸和磷酸,进一步转变成6-磷酸葡萄糖,最终转变为葡萄糖或糖原。此外,磷酸二羟丙酮还可通过糖酵解-氧化途径转变成乙酰辅酶A,从而合成脂肪果糖,在上述酶的作用下最终约有50%转化为葡萄糖,其余则生成糖原丙酮酸、三酸甘油酯和脂肪等。遗传性果糖不耐症是由于1-磷酸果糖醛缩酶(fructose-1-P aldolase)缺陷所致。已知该酶的分子量为16万,由4个亚单位组成;根据其催化活性免疫特征和在不同组织中的分布情况又可分为A、B、C三型同工酶,在肝肾和小肠中以B型果糖二磷酸醛缩酶为主,它的编码基因位于9q13~q32,长约14500bp。
研究结果显示A149P、A174D和N334k三种点突变是导致果糖不耐症的最主要原因。本病患儿肝脏内的1-磷酸果糖醛缩酶活性由完全缺乏到仅为正常人的12%左右不等,由于该酶缺乏果糖代谢的肝和肾中都有1-磷酸果糖堆积,除对细胞有损害外,过多的1-磷酸果糖不仅可使果糖-1,6-二磷酸酶活性受到抑制,还使从甘油、氨基酸等转变成葡萄糖的糖原异生作用受阻,从而引起低血糖,此外过多的1-磷酸果糖还抑制磷酸化酶的作用,阻碍糖原转变成葡萄糖,这是导致低血糖的另一原因,而且由于大量无机磷亦同时被消耗,使得血磷降低和ATP再生减少。1-磷酸果糖的累积和ATP供应不足也阻碍了糖原转换成1-磷酸葡萄糖,如继续使用含蔗糖或果糖的食物喂养,将造成患儿肝细胞损伤,持久的含果糖饮食会造成患儿肝细胞坏死、脂肪浸润、胆小管增生和纤维化,甚至肝硬化,其机制还不明确,可能是由于1-磷酸果糖的细胞毒性作用或与缺乏ATP有关。
通过对果糖不耐受症患者的家系进行研究,已发现ALDOB基因的16个不同的突变位点,最主要的突变发现在A149P(64%)、A174D(16%)以及N335K(5%),单倍体分析证明A149P和A174D ALDOB突变体起始于单一祖先,通过遗传漂移目前已经达到了一个相当高的突变频率,在英国,大约有1.3%的新生儿有A149P基因突变。







